-
INTRODUCTION
This is a web-based implementation for calculation of Sterimol parameters for organic molecules, developed by Tipker and Verloop (Pesticide Synthesis Through Rational Approaches, 1984, Chapter 16, pp 279-296, DOI: 10.1021/bk-1984-0255.ch016). Web version was inspired by wSterimol, Sterimol and DBSTEP command-line python programs developed by Brethomé and Paton (ACS Catal., 2019, 9(3), pp 2313–2323, DOI: 10.1021/acscatal.8b04043, GitHub, Zenodo DOI:10.5281/zenodo.1434440), and Luchini, Patterson, and Paton (Zenodo DOI:10.5281/zenodo.4702097). The main difference is the usage of xyz coordinates (in .xyz), calculation on a complete system, no requirement for installation procedure, and direct visualization of Sterimol sub-parameters (L0, B1, and B5).
Additionally, burried volume (%VBur) is calculated using similar aproach as SambVca 2.1, developed by Cavallo et al (Nature Chem., 2019, 11, pp. 872–879, DOI:10.1038/s41557-019-0319-5). For calculation of buried volume only definition of reaction centre is needed (see Input section). Ignoring reaction centre radius, target radius of 3.5 ångströms, mesh spacing value of 0.1 ångströms, and Bondi atomic radii scaled by 1.17 are used for the calculation.
INPUT
For input, a modified XYZ file is needed. XYZ format specifies the molecule geometry by giving the number of atoms with Cartesian coordinates that will be read on the first line, a comment on the second (usually name and/or energy), and the lines of atomic coordinates in the following lines. The units are in ångströms. After definition of atom type, x, y, and z coordinates, additional column Frag is used for assignation of fragments (0 for fixed, 1 for variable part (-R)), while column Atrib assigns atoms for primary bond of Sterimol (L0 parameter (1 and 2)) and target reaction centre (3) for SambVca %VBur. For analysis of multiple conformations, previously generated .xyz and .trj (from xtb and ORCA programs), with the same ordering of atoms as in input coordinates should be selected in the drop-zone (choose file). Botzmann averaging is performed at 298.15 K.
Example file for multimolecule file can be found here.
CREDITS
This software was developed by Davor Šakić and Gabrijel Zubčić. Help from Valerije Vrček (discussion and testing) and Mateja Toma (testing) is greatly appreciated.
Funding: HrZZ, OrDeN, LIGHT-N-RING, 2022.
Institution: University of Zagreb, Faculty of Pharmacy and Biochemistry


DISCLAIMER
The software and results are provided "AS IS", without warranty of any kind, express or implied. In no event shall the authors or copyright holders be liable for any claim, damages or other liability. Good luck! -
As used in:
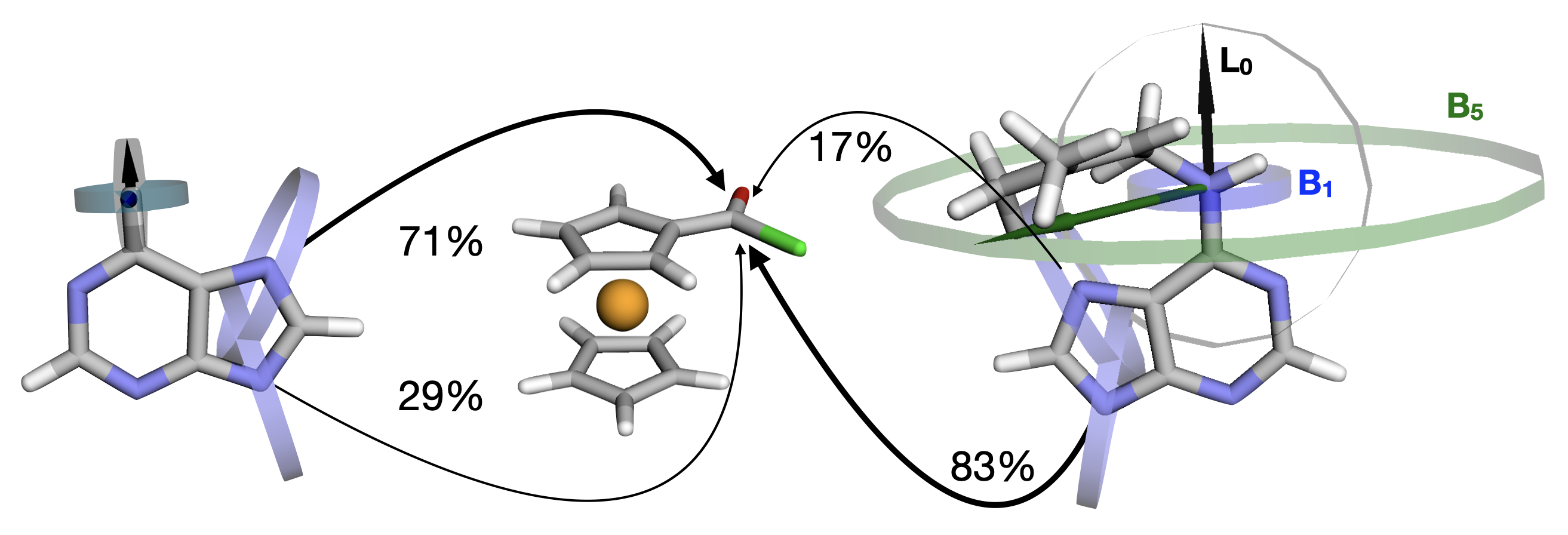
Toma, Mateja; Zubčić, Gabrijel; Lapić, Jasmina; Djaković, Senka; Šakić, Davor; Vrček, Valerije
Ferrocenoyl-adenines: substituent effects on regioselective acylation
Beilstein journal of organic chemistry, 2022, 18, 1270–1277
DOI: 10.3762/bjoc.18.133
-
INPUT COORDINATES
Allow few minutes for calculation of
all parameters in multimolecule file
-
SteriXYZ with SambVca
-
Powered by: